|
|
|
|
|
|
|

|
|
|
|

|
|
|
|
|
|
|
|
## User Guide (Version 0.1)
|
|
|
|
|
| ... | ... | @@ -9,117 +8,74 @@ Gaurav Saxena, Miguel Ponce-de-Leon, Arnau Montagud,David Vicente Dorca and Alfo |
|
|
|
|
|
|
|
Revision: _16 March, 2022_
|
|
|
|
|
|
|
|
|
|
|
|
[[_TOC_]]
|
|
|
|
* [User Guide (Version 0.1)](#user-guide-version-01)
|
|
|
|
* [Introduction](#introduction)
|
|
|
|
* [The Current Version](#the-current-version)
|
|
|
|
* [Layers in PhysiCell-X](#layers-in-physicell-x)
|
|
|
|
* [Parallel Macroscopic Design](#parallel-macroscopic-design)
|
|
|
|
* [An Analogy](#an-analogy)
|
|
|
|
* [Domain Partitioning](#domain-partitioning)
|
|
|
|
* [Mapping to Hardware](#mapping-to-hardware)
|
|
|
|
* [Prerequisites](#prerequisites)
|
|
|
|
* [MPI processes, Voxels and Divisibility](#mpi-processes-voxels-and-divisibility)
|
|
|
|
* [Code-base organization](#code-base-organization)
|
|
|
|
* [Running an Example](#running-an-example)
|
|
|
|
* [Running the TNF MPI example](#running-the-tnf-mpi-example)
|
|
|
|
* [Future Work](#future-work)
|
|
|
|
* [Acknowledgements](#acknowledgements)
|
|
|
|
* [Footnotes:](#footnotes)
|
|
|
|
* [References:](#references)
|
|
|
|
|
|
|
|
# Introduction
|
|
|
|
|
|
|
|
|
|
|
|
PhysiCell-X is the distributed version of PhysiCell [2].
|
|
|
|
PhysiCell is a open-source, multi-physics, multi-scale, agent-based simulator for biological systems.
|
|
|
|
It provides both the stage (micro-environment) and actors (cells or agents) for simulation.
|
|
|
|
Though PhysiCell is light-weight, flexible and shared-memory parallelized using OpenMP (Open Multiprocessing), it *cannot* run on distributed systems i.e. it cannot be executed on multiple nodes of an HPC (High Performance Computing) cluster.
|
|
|
|
Thus, the problem size that PhysiCell can handle is limited by the maximum memory of a single node.
|
|
|
|
This is a limitation that needs to be removed and this is where PhysiCell-X comes in.
|
|
|
|
PhysiCell-X enables the distributed parallelization of PhysiCell by making use of MPI (Message-Passing Interface). In simple words, you can now use multiple nodes of an HPC system to solve a single, coherent problem.
|
|
|
|
Thus, the aim of PhysiCell-X is to remove the memory limitation, reduce the time to solution by distributing the computation onto multiple compute nodes and solve very large sized (real-world scale) problems.
|
|
|
|
PhysiCell-X is the distributed version of PhysiCell \[2\]. PhysiCell is a open-source, multi-physics, multi-scale, agent-based simulator for biological systems. It provides both the stage (micro-environment) and actors (cells or agents) for simulation. Though PhysiCell is light-weight, flexible and shared-memory parallelized using OpenMP (Open Multiprocessing), it _cannot_ run on distributed systems i.e. it cannot be executed on multiple nodes of an HPC (High Performance Computing) cluster. Thus, the problem size that PhysiCell can handle is limited by the maximum memory of a single node. This is a limitation that needs to be removed and this is where PhysiCell-X comes in. PhysiCell-X enables the distributed parallelization of PhysiCell by making use of MPI (Message-Passing Interface). In simple words, you can now use multiple nodes of an HPC system to solve a single, coherent problem. Thus, the aim of PhysiCell-X is to remove the memory limitation, reduce the time to solution by distributing the computation onto multiple compute nodes and solve very large sized (real-world scale) problems.
|
|
|
|
|
|
|
|
## The Current Version
|
|
|
|
|
|
|
|
The current version of PhysiCell-X is 0.1 and it is based on PhysiCell version 1.9.0. Please note the following very carefully.
|
|
|
|
|
|
|
|
- The User Guide for PhysiCell-X is to be used in conjunction with the User Guide for PhysiCell (till version 1.9.0). The User Guide for PhysiCell contains many details which are *not* reproduced here to avoid unnecessary duplication.
|
|
|
|
|
|
|
|
- PhysiCell-X *only* focuses on 3-D problems and hence distributed parallelism is available *only* for 3-D problems.
|
|
|
|
|
|
|
|
- PhysiCell-X uses MPI for distributed parallelization and OpenMP shared-memory parallelization was already available in PhysiCell. Thus, PhysiCell actually uses hybrid parallelization i.e. MPI + OpenMP.
|
|
|
|
|
|
|
|
- A (very) small set of features available in PhysiCell version 1.9.0 are *not* available in PhysiCell-X. We will list out all such functionalities in a *separate section*. As the future versions of PhysiCell-X are released, we will try our best to incorporate these features.
|
|
|
|
* The User Guide for PhysiCell-X is to be used in conjunction with the User Guide for PhysiCell (till version 1.9.0). The User Guide for PhysiCell contains many details which are _not_ reproduced here to avoid unnecessary duplication.
|
|
|
|
* PhysiCell-X _only_ focuses on 3-D problems and hence distributed parallelism is available _only_ for 3-D problems.
|
|
|
|
* PhysiCell-X uses MPI for distributed parallelization and OpenMP shared-memory parallelization was already available in PhysiCell. Thus, PhysiCell actually uses hybrid parallelization i.e. MPI + OpenMP.
|
|
|
|
* A (very) small set of features available in PhysiCell version 1.9.0 are _not_ available in PhysiCell-X. We will list out all such functionalities in a _separate section_. As the future versions of PhysiCell-X are released, we will try our best to incorporate these features.
|
|
|
|
|
|
|
|
> **IMPORTANT**
|
|
|
|
|
|
|
|
> Please use *this* User Guide in *conjunction* with the User Guide for PhysiCell (till version 1.9.0). The User Guide for PhysiCell contains several details which are *not* reproduced here.
|
|
|
|
> Please use _this_ User Guide in _conjunction_ with the User Guide for PhysiCell (till version 1.9.0). The User Guide for PhysiCell contains several details which are _not_ reproduced here.
|
|
|
|
|
|
|
|
## Layers in PhysiCell-X
|
|
|
|
|
|
|
|
PhysiCell-X can be visualized as being made up of two layers.
|
|
|
|
The first layer BioFVM-X [4] solves the diffusion equations of substrates in the micro-environment.
|
|
|
|
The second layer is PhysiCell-X itself and takes care of the movement, growth, decay, division, chemical and mechanical interaction and death of cells (agents) etc. BioFVM-X has been released separately and is available at:
|
|
|
|
PhysiCell-X can be visualized as being made up of two layers. The first layer BioFVM-X \[4\] solves the diffusion equations of substrates in the micro-environment. The second layer is PhysiCell-X itself and takes care of the movement, growth, decay, division, chemical and mechanical interaction and death of cells (agents) etc. BioFVM-X has been released separately and is available at:
|
|
|
|
|
|
|
|
1. [Zenodo](https://zenodo.org/record/5044998#.YfjVwi8RppQ) and
|
|
|
|
|
|
|
|
2. [GitLab](https://gitlab.bsc.es/gsaxena/biofvm_x) (a detailed tutorial $^1$ to run examples can be found here)
|
|
|
|
|
|
|
|
The latest version of BioFVM-X also forms part of PhysiCell-X.
|
|
|
|
It can kindly be noted that the stand-alone version of BioFVM-X available at the links above is *different* from the version of BioFVM-X that comes bundled with PhysiCell-X.
|
|
|
|
This is because the design of BioFVM-X has evolved as PhysiCell-X evolved.
|
|
|
|
The latest version of BioFVM-X also forms part of PhysiCell-X. It can kindly be noted that the stand-alone version of BioFVM-X available at the links above is _different_ from the version of BioFVM-X that comes bundled with PhysiCell-X. This is because the design of BioFVM-X has evolved as PhysiCell-X evolved.
|
|
|
|
|
|
|
|
## Parallel Macroscopic Design
|
|
|
|
|
|
|
|
### An Analogy
|
|
|
|
|
|
|
|
We describe the high level parallel design with the help of a non-technical analogy of a "Partitioned Aquarium".
|
|
|
|
Imagine a large aquarium in the shape of a cuboid. It is filled with fish, water, nutrients that we use to feed the fish and everything that you can imagine in an aquarium.
|
|
|
|
The aquarium is equivalent to our *3-D domain*.
|
|
|
|
The fish which can grow, move, die, ingest/secrete nutrients, interact with other fish and so on are equivalent to the *cells* or *agents*.
|
|
|
|
The water and the dissolved/undissolved nutrients and other things present are like the *micro-environment*.
|
|
|
|
Now, further imagine that we place fictitious partitions in the aquarium which do not let the water/nutrients from one sub-part move to another sub-part but the fish can move from one sub-part to another *adjacent* sub-part through these partitions.
|
|
|
|
These sub-partitions of the aquarium are equivalent to our sub-domains.
|
|
|
|
Thus, the micro-environment in a sub-domain cannot move to the adjacent sub-domain *but* the cells or agents *can* move to *adjacent* sub-domains.
|
|
|
|
Further, instead of keeping a single person to manage the aquarium, we hire one person each to manage each of the sub-partitions.
|
|
|
|
Each of these persons *independently* manages one sub-partition.
|
|
|
|
A person here is equivalent to an *Operating System process* (more specifically an *MPI process*) and the sub-partition is equivalent to a sub-domain.
|
|
|
|
To take things further, each of these persons can hire additional people under them to manage the sub-partition.
|
|
|
|
These *additional* people are like *OpenMP threads* which run within an MPI process.
|
|
|
|
We describe the high level parallel design with the help of a non-technical analogy of a "Partitioned Aquarium". Imagine a large aquarium in the shape of a cuboid. It is filled with fish, water, nutrients that we use to feed the fish and everything that you can imagine in an aquarium. The aquarium is equivalent to our _3-D domain_. The fish which can grow, move, die, ingest/secrete nutrients, interact with other fish and so on are equivalent to the _cells_ or _agents_. The water and the dissolved/undissolved nutrients and other things present are like the _micro-environment_. Now, further imagine that we place fictitious partitions in the aquarium which do not let the water/nutrients from one sub-part move to another sub-part but the fish can move from one sub-part to another _adjacent_ sub-part through these partitions. These sub-partitions of the aquarium are equivalent to our sub-domains. Thus, the micro-environment in a sub-domain cannot move to the adjacent sub-domain _but_ the cells or agents _can_ move to _adjacent_ sub-domains. Further, instead of keeping a single person to manage the aquarium, we hire one person each to manage each of the sub-partitions. Each of these persons _independently_ manages one sub-partition. A person here is equivalent to an _Operating System process_ (more specifically an _MPI process_) and the sub-partition is equivalent to a sub-domain. To take things further, each of these persons can hire additional people under them to manage the sub-partition. These _additional_ people are like _OpenMP threads_ which run within an MPI process.
|
|
|
|
|
|
|
|
### Domain Partitioning
|
|
|
|
|
|
|
|
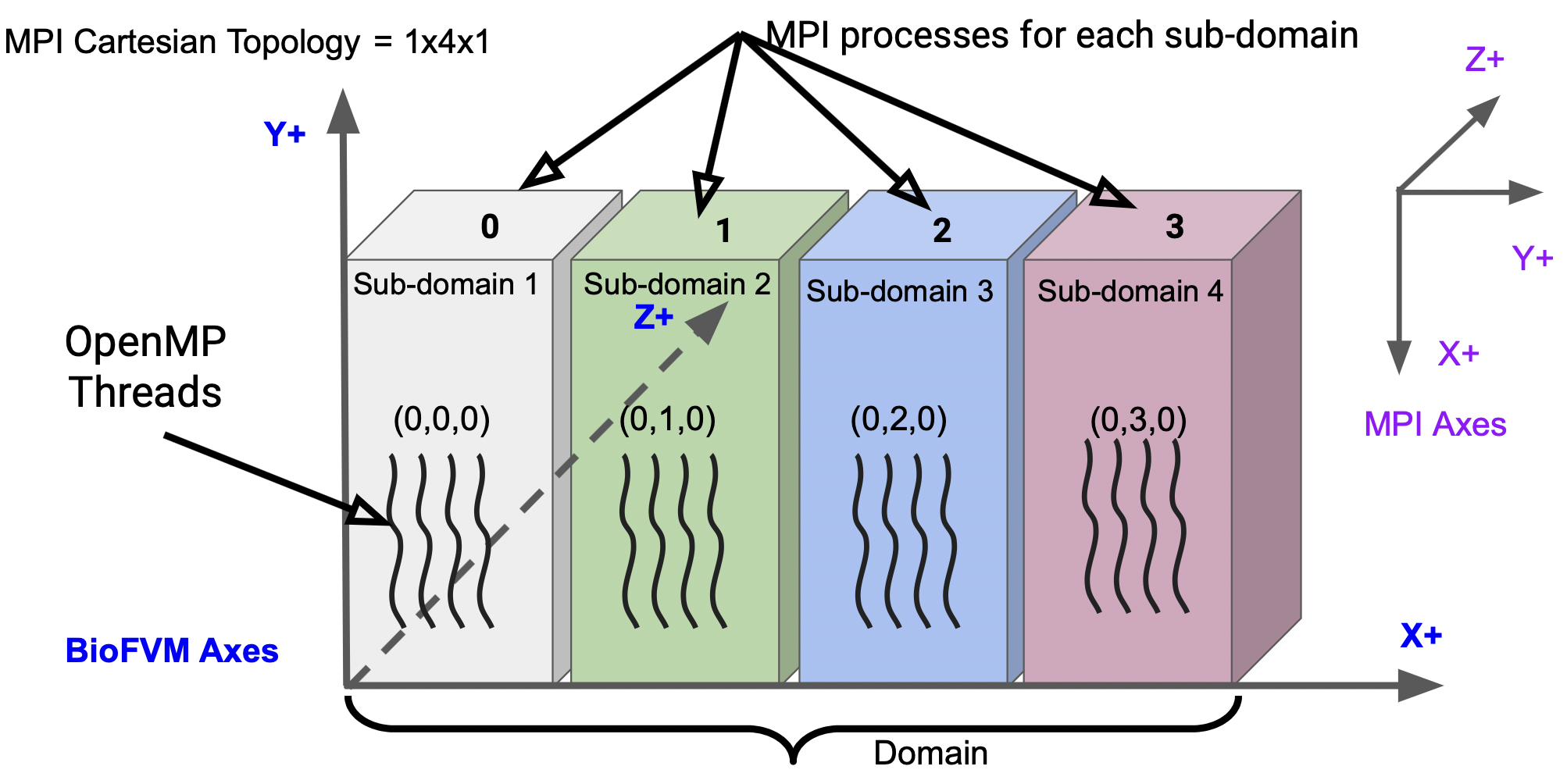
Figure [\[fig:domain\_partitioning\]](#fig:domain_partitioning){reference-type="ref" reference="fig:domain_partitioning"} formally illustrates the aforementioned analogy.
|
|
|
|
It shows a 3-D domain and the directions of the axes.
|
|
|
|
The domain is divided in the X-direction *only* among the MPI processes i.e. the 3-D domain is partitioned in a single dimension only (1-D domain partitioning - imagine slices of a bread).
|
|
|
|
It is important to note that the direction of the axes of BioFVM-X/PhysiCell-X is different from the directions of the axes of MPI Cartesian Topology $^2$.
|
|
|
|
In this specific case, the whole 3-D domain is partitioned among 4 MPI processes (shown in gray, green, blue and red). This Cartesian Topology is $1 \times 4 \times 1$, indicating that we have 1 MPI process in the X-direction, 4 in the Y-direction and 1 in the Z-direction.
|
|
|
|
Please note we have 4 processes in the Y-direction of the MPI Cartesian Topology because *the X-axis of BioFVM-X/PhysiCell-X is equivalent to the Y-axis of the MPI Topology*.
|
|
|
|
Each of these sub-partitions (formally called sub-domains) can be located with the help of MPI Cartesian coordinates.
|
|
|
|
Process 0 (formally called Rank 0) has coordinates $(0,0,0)$, process 1 (Rank 1) has $(0,1,0)$, process 2 (Rank 2) has $(0,2,0)$ and process 4 (Rank 3) has coordinates $(0,3,0)$.
|
|
|
|
Within each sub-domain managed by a single MPI process, the wavy, dark, solid lines indicate OpenMP threads.
|
|
|
|
Figure [\[fig:domain_partitioning\]](#fig:domain_partitioning){reference-type="ref" reference="fig:domain_partitioning"} formally illustrates the aforementioned analogy. It shows a 3-D domain and the directions of the axes. The domain is divided in the X-direction _only_ among the MPI processes i.e. the 3-D domain is partitioned in a single dimension only (1-D domain partitioning - imagine slices of a bread). It is important to note that the direction of the axes of BioFVM-X/PhysiCell-X is different from the directions of the axes of MPI Cartesian Topology $^2$. In this specific case, the whole 3-D domain is partitioned among 4 MPI processes (shown in gray, green, blue and red). This Cartesian Topology is $1 \\times 4 \\times 1$, indicating that we have 1 MPI process in the X-direction, 4 in the Y-direction and 1 in the Z-direction. Please note we have 4 processes in the Y-direction of the MPI Cartesian Topology because _the X-axis of BioFVM-X/PhysiCell-X is equivalent to the Y-axis of the MPI Topology_. Each of these sub-partitions (formally called sub-domains) can be located with the help of MPI Cartesian coordinates. Process 0 (formally called Rank 0) has coordinates $(0,0,0)$, process 1 (Rank 1) has $(0,1,0)$, process 2 (Rank 2) has $(0,2,0)$ and process 4 (Rank 3) has coordinates $(0,3,0)$. Within each sub-domain managed by a single MPI process, the wavy, dark, solid lines indicate OpenMP threads.
|
|
|
|
|
|
|
|
### Mapping to Hardware
|
|
|
|
|
|
|
|
Each sub-domain has a different and independent micro-environment which
|
|
|
|
is managed by a single MPI process. Multiple OpenMP threads can exist
|
|
|
|
within each MPI process. *Typically*, to obtain a good performance from
|
|
|
|
PhysiCell-X, one would run a single MPI process per socket and one
|
|
|
|
OpenMP thread per core. For example, if a node of an HPC cluster
|
|
|
|
consists of 2 sockets and each socket has 24 cores then we *typically*
|
|
|
|
would run 2 MPI processes per node i.e. one MPI process per socket and
|
|
|
|
then run 24 OpenMP threads per socket i.e. one OpenMP thread per
|
|
|
|
hardware core. Please note that this is *not* a rule and sometimes we
|
|
|
|
also run 1 MPI process per node and 48 OpenMP threads per MPI process.
|
|
|
|
Each sub-domain has a different and independent micro-environment which is managed by a single MPI process. Multiple OpenMP threads can exist within each MPI process. _Typically_, to obtain a good performance from PhysiCell-X, one would run a single MPI process per socket and one OpenMP thread per core. For example, if a node of an HPC cluster consists of 2 sockets and each socket has 24 cores then we _typically_ would run 2 MPI processes per node i.e. one MPI process per socket and then run 24 OpenMP threads per socket i.e. one OpenMP thread per hardware core. Please note that this is _not_ a rule and sometimes we also run 1 MPI process per node and 48 OpenMP threads per MPI process.
|
|
|
|
|
|
|
|

|
|
|
|
|
|
|
|
|
|
|
|
## Prerequisites
|
|
|
|
|
|
|
|
PhysiCell-X is completely written in C++ and uses MPI + OpenMP for distributed-shared memory parallelization. There is no need for a package manager and can be installed using Makefiles. Thus there are two prerequisites:
|
|
|
|
|
|
|
|
PhysiCell-X is completely written in C++ and uses MPI + OpenMP for
|
|
|
|
distributed-shared memory parallelization. There is no need for a
|
|
|
|
package manager and can be installed using Makefiles. Thus there are two
|
|
|
|
prerequisites:
|
|
|
|
1. A C++ compiler with a support for OpenMP for e.g., the GCC g++ compiler, the Intel C++, or Apple clang compiler etc.
|
|
|
|
2. An MPI implementation for e.g., OpenMPI, MPICH, Mvapich2, Intel MPI, IBM Spectrum MPI or others.
|
|
|
|
|
|
|
|
1. A C++ compiler with a support for OpenMP for e.g., the GCC g++
|
|
|
|
compiler, the Intel C++, or Apple clang compiler etc.
|
|
|
|
|
|
|
|
2. An MPI implementation for e.g., OpenMPI, MPICH, Mvapich2, Intel MPI,
|
|
|
|
IBM Spectrum MPI or others.
|
|
|
|
|
|
|
|
Please see the original User documentation of PhysiCell for instructions
|
|
|
|
on how to install on OSX (MacOS) and Virtual Machine (like VirtualBox).
|
|
|
|
Some MATLAB scripts for plotting are bundled with PhysiCell/PhysiCell-X
|
|
|
|
which can be executed in MATLAB or *possibly*
|
|
|
|
[Octave](https://www.gnu.org/software/octave/). We also recommend
|
|
|
|
installing [ImageMagick](http://imagemagick.org) which is useful for
|
|
|
|
converting SVG images to PNG, JPEG or creating a movie from SVG files.
|
|
|
|
Please see the original User documentation of PhysiCell for instructions on how to install on OSX (MacOS) and Virtual Machine (like VirtualBox). Some MATLAB scripts for plotting are bundled with PhysiCell/PhysiCell-X which can be executed in MATLAB or _possibly_ [Octave](https://www.gnu.org/software/octave/). We also recommend installing [ImageMagick](http://imagemagick.org) which is useful for converting SVG images to PNG, JPEG or creating a movie from SVG files.
|
|
|
|
|
|
|
|
> **📝 IMPORTANT**
|
|
|
|
|
| ... | ... | @@ -127,216 +83,177 @@ converting SVG images to PNG, JPEG or creating a movie from SVG files. |
|
|
|
|
|
|
|
# MPI processes, Voxels and Divisibility
|
|
|
|
|
|
|
|
The 3-D simulation domain in PhysiCell-X is divided into Voxels
|
|
|
|
(Volumetric Pixels). Voxels generally are cubic (but they can be a
|
|
|
|
cuboid as well). Thus, for example if the domain length in the X, Y and
|
|
|
|
Z direction is \[-500,+500\], \[-500,+500\] and \[-500,+500\],
|
|
|
|
respectively and the Voxel (cubic) dimension is 20, then we have
|
|
|
|
$\frac{500 - (-500)}{25} = \frac{1000}{20}=50$ voxels *each* in the X, Y
|
|
|
|
and Z-directions. As described in the previous section, PhysiCell-X
|
|
|
|
implements 1-D domain partitioning in the X-direction and due to a
|
|
|
|
limitation in our implementation (will be removed in future versions),
|
|
|
|
the total number of Voxels in the X-direction should be perfectly
|
|
|
|
divisible by the total number of MPI processes. Thus, in the case above,
|
|
|
|
we cannot have 3 MPI processes as 3 does not perfectly divide 50.
|
|
|
|
However, we can have 2, 10, or even 25 MPI processes as these divide 50
|
|
|
|
voxels perfectly. It can be noted that this limitation exists *only* in
|
|
|
|
the X-direction and *not* in the Y or Z directions. There is no
|
|
|
|
restriction on the number of OpenMP threads within a single MPI process.
|
|
|
|
Further, there are two types of meshes in PhysiCell (or PhysiCell-X)
|
|
|
|
i.e. a *diffusion mesh* and a *mechanical mesh*. The size of the voxels
|
|
|
|
for *each* of these meshes is defined separately. It is important that
|
|
|
|
the perfect divisibility condition should hold for *both* the types of
|
|
|
|
meshes. We will return to this concept when we show how to run examples
|
|
|
|
and specify parameters in PhysiCell-X.
|
|
|
|
The 3-D simulation domain in PhysiCell-X is divided into Voxels (Volumetric Pixels). Voxels generally are cubic (but they can be a cuboid as well). Thus, for example if the domain length in the X, Y and Z direction is \[-500,+500\], \[-500,+500\] and \[-500,+500\], respectively and the Voxel (cubic) dimension is 20, then we have $\\frac{500 - (-500)}{25} = \\frac{1000}{20}=50$ voxels _each_ in the X, Y and Z-directions. As described in the previous section, PhysiCell-X implements 1-D domain partitioning in the X-direction and due to a limitation in our implementation (will be removed in future versions), the total number of Voxels in the X-direction should be perfectly divisible by the total number of MPI processes. Thus, in the case above, we cannot have 3 MPI processes as 3 does not perfectly divide 50. However, we can have 2, 10, or even 25 MPI processes as these divide 50 voxels perfectly. It can be noted that this limitation exists _only_ in the X-direction and _not_ in the Y or Z directions. There is no restriction on the number of OpenMP threads within a single MPI process. Further, there are two types of meshes in PhysiCell (or PhysiCell-X) i.e. a _diffusion mesh_ and a _mechanical mesh_. The size of the voxels for _each_ of these meshes is defined separately. It is important that the perfect divisibility condition should hold for _both_ the types of meshes. We will return to this concept when we show how to run examples and specify parameters in PhysiCell-X.
|
|
|
|
|
|
|
|
> **📝 IMPORTANT**
|
|
|
|
>
|
|
|
|
> 1. The total number of voxels in the X-direction must be perfectly divisible by the total number of MPI processes.
|
|
|
|
> 2. Condition 1 above applies to both Diffusion and Mechanical voxels.
|
|
|
|
> 3. Size of Diffusion voxel must be $\leq$ size of Mechanical voxel.
|
|
|
|
> 3. Size of Diffusion voxel must be $\\leq$ size of Mechanical voxel.
|
|
|
|
|
|
|
|
# Code-base organization
|
|
|
|
|
|
|
|
Inside the parent directory of PhysiCell-X, there are multiple
|
|
|
|
directories. Some directories which users would frequently need to deal
|
|
|
|
with are:
|
|
|
|
|
|
|
|
- `config:` This directory is used for providing the inputs to the application. We use a file named `PhysiCell_settings.xml` to provide *most* of the input parameters. We say *most* because there are some parameters that *can* be provided in the file that contains the `main()` function of C++ (whatever that file is named).
|
|
|
|
Inside the parent directory of PhysiCell-X, there are multiple directories. Some directories which users would frequently need to deal with are:
|
|
|
|
|
|
|
|
- `custom_modules:` This directory is where the custom code created by the user is put that is specific to an example that the user is working with. *Typically*, the file is named `PhysiCell_custom.cpp` and `PhysiCell_custom.h`.
|
|
|
|
* `config:` This directory is used for providing the inputs to the application. We use a file named `PhysiCell_settings.xml` to provide _most_ of the input parameters. We say _most_ because there are some parameters that _can_ be provided in the file that contains the `main()` function of C++ (whatever that file is named).
|
|
|
|
* `custom_modules:` This directory is where the custom code created by the user is put that is specific to an example that the user is working with. _Typically_, the file is named `PhysiCell_custom.cpp` and `PhysiCell_custom.h`.
|
|
|
|
* `sample_projects` _and_ `sample_projects_intracellular:` These directories includes sample projects that can be used as a starting point to create your own example. Users can _check-out_ a project from these directories into the _working_ directories and work with the copy of the checked-out code. The strategy of working with a _copy_ of the code prevents any accidental/unwanted changes into the original code. Once the user is satisfied with the _working copy_, this can be checked back into the `sample_projects` or `sample_projects_intracellular` master code.
|
|
|
|
|
|
|
|
- `sample_projects` *and* `sample_projects_intracellular:` These directories includes sample projects that can be used as a starting point to create your own example. Users can *check-out* a project from these directories into the *working* directories and work with the copy of the checked-out code. The strategy of working with a *copy* of the code prevents any accidental/unwanted changes into the original code. Once the user is satisfied with the *working copy*, this can be checked back into the `sample_projects` or `sample_projects_intracellular` master code.
|
|
|
|
|
|
|
|
There are multiple other directories which *typically* a user will not need to interact with. Some directories are:
|
|
|
|
|
|
|
|
1. `BioFVM:` This includes a working copy of the BioFVM/BioFVM-X multi-substrate diffusion code [2; 4]. BioFVM distributions also include pugixml (an efficient cross-platform XML parser) [3].
|
|
|
|
There are multiple other directories which _typically_ a user will not need to interact with. Some directories are:
|
|
|
|
|
|
|
|
1. `BioFVM:` This includes a working copy of the BioFVM/BioFVM-X multi-substrate diffusion code \[2; 4\]. BioFVM distributions also include pugixml (an efficient cross-platform XML parser) \[3\].
|
|
|
|
2. `core:` contains the core library files for PhysiCell/PhysiCell-X. The `modules` directory also contains some core files. In the future the developers plan to remove the `modules` directory.
|
|
|
|
|
|
|
|
3. `matlab` This includes basic Matlab files for handling PhysiCell outputs. These can be used to visualize the output data.
|
|
|
|
|
|
|
|
> **📝 IMPORTANT: Note on the parallel code**:
|
|
|
|
|
|
|
|
> Whenever the parallel equivalent of a serial function is available, it is written immediately below the corresponding serial function in the same source file.
|
|
|
|
|
|
|
|
|
|
|
|
# Running an Example
|
|
|
|
|
|
|
|
After downloading PhysiCell-X, go to the parent/top-level
|
|
|
|
directory/folder. As of now, there are three examples that are hybrid
|
|
|
|
parallelized (MPI+OpenMP). These are:
|
|
|
|
After downloading PhysiCell-X, go to the parent/top-level directory/folder. As of now, there are three examples that are hybrid parallelized (MPI+OpenMP). These are:
|
|
|
|
|
|
|
|
1. `sample_projects/pred_prey_mpi`: This is a classic Predator-Prey project in 3-D.
|
|
|
|
|
|
|
|
2. `sample_projects/heterogeneity_mpi`: simulates a 3-D tumor with heterogeneous "genetics" that drive differential proliferation [2] (text reproduced from the original PhysiCell User Guide but instead of a 2-D tumor, we now have a 3-D tumor).
|
|
|
|
|
|
|
|
2. `sample_projects/heterogeneity_mpi`: simulates a 3-D tumor with heterogeneous "genetics" that drive differential proliferation \[2\] (text reproduced from the original PhysiCell User Guide but instead of a 2-D tumor, we now have a 3-D tumor).
|
|
|
|
3. `sample_projects_intracellular/boolean/spheroid_tnf_model_mpi`: This is a paralleized example in 3-D which makes use of PhysiBoss (bundled with PhysiCell v1.9.0/PhysiCell-X v0.1) to simulate the Tumor Necrosis Factor (TNF) simulation.
|
|
|
|
|
|
|
|
The first step is to *check-out/populate* a project. Checking
|
|
|
|
out/populating means *copying* a project and working files from the
|
|
|
|
project directory into the working directory. Thus, as mentioned before,
|
|
|
|
working with a copy of the projects prevents the original copy from
|
|
|
|
being modified. This also has the *disadvantage* that modified files
|
|
|
|
need to be copied to the original project directories if you are
|
|
|
|
satisfied with the experiment and changes. To populate a project:
|
|
|
|
The first step is to _check-out/populate_ a project. Checking out/populating means _copying_ a project and working files from the project directory into the working directory. Thus, as mentioned before, working with a copy of the projects prevents the original copy from being modified. This also has the _disadvantage_ that modified files need to be copied to the original project directories if you are satisfied with the experiment and changes. To populate a project:
|
|
|
|
|
|
|
|
make [project_name]
|
|
|
|
```plaintext
|
|
|
|
make [project_name]
|
|
|
|
```
|
|
|
|
|
|
|
|
where the `project_name` is one of the following three:
|
|
|
|
|
|
|
|
1. `pred-prey-mpi`
|
|
|
|
|
|
|
|
2. `heterogeneity-sample-mpi`
|
|
|
|
|
|
|
|
3. `physiboss-tnf-model-mpi`
|
|
|
|
|
|
|
|
> **📝 IMPORTANT: Note on the `make` function**:
|
|
|
|
> 1. To see all sample projects type `make list-projects`. The name of the project can be different from the name of the directory under which it is stored. Name of the project *typically* contains *hyphens* whereas the project directory name contains *underscores*.
|
|
|
|
> **📝 IMPORTANT: Note on the** `make` function:
|
|
|
|
>
|
|
|
|
> 1. To see all sample projects type `make list-projects`. The name of the project can be different from the name of the directory under which it is stored. Name of the project _typically_ contains _hyphens_ whereas the project directory name contains _underscores_.
|
|
|
|
> 2. To clear object files, executable files of a project type `make clean`.
|
|
|
|
> 3. To reset the PhysiCell-X to the original (or clean) state type `make reset` (remember to copy any modified file *into* the appropriate project before you execute this command and also remove any unwanted remaining files in the `config` directory !)
|
|
|
|
> 3. To reset the PhysiCell-X to the original (or clean) state type `make reset` (remember to copy any modified file _into_ the appropriate project before you execute this command and also remove any unwanted remaining files in the `config` directory !)
|
|
|
|
|
|
|
|
Checking-out a project will first:
|
|
|
|
|
|
|
|
1. Replace the `Makefile` in the parent directory with the *project-specific* `Makfile`
|
|
|
|
|
|
|
|
1. Replace the `Makefile` in the parent directory with the _project-specific_ `Makfile`
|
|
|
|
2. Copy the `main.cpp` (file name may vary with project but this file would contain the C++ `main()` function) of the project to the working parent directory.
|
|
|
|
|
|
|
|
3. Copy the *project-specific* files in the `custom_modules` directory in the `custom_modules` directory under the parent directory.
|
|
|
|
|
|
|
|
3. Copy the _project-specific_ files in the `custom_modules` directory in the `custom_modules` directory under the parent directory.
|
|
|
|
4. Copy the project specific files in the `config` directory to `config` under the parent directory.
|
|
|
|
|
|
|
|
**📝 IMPORTANT: Note on `PhysiCell settings.xml`**:
|
|
|
|
There can be multiple `PhysiCell_settings_.xml` file inside the `config` directory. *Typically* it is the plain, simple file named `PhysiCell_settings.xml` that the user can start with.
|
|
|
|
> **📝 IMPORTANT: Note on** `PhysiCell settings.xml`
|
|
|
|
>
|
|
|
|
> There can be multiple `PhysiCell_settings_.xml` file inside the `config` directory. _Typically_ it is the plain, simple file named `PhysiCell_settings.xml` that the user can start with.
|
|
|
|
|
|
|
|
As an example, assume that the parent directory of PhysiCell-X is `PhysiCell-X` and we need to check-out the `pred-prey-mpi` project inside the `sample_projects` directory. On executing
|
|
|
|
|
|
|
|
make pred-prey-mpi
|
|
|
|
```plaintext
|
|
|
|
make pred-prey-mpi
|
|
|
|
```
|
|
|
|
|
|
|
|
the following happens:
|
|
|
|
|
|
|
|
1. Copy file `PhysiCell-X/sample_projects/pred_prey_mpi/Makefile` to `PhysiCell-X/Makefile`.
|
|
|
|
|
|
|
|
2. Copy file `PhysiCell-X/sample_projects/pred_prey_mpi/main.cpp` to `Physicell-X/main.cpp`
|
|
|
|
|
|
|
|
3. Copy directory `Physicell-X/sample_projects/pred_prey_mpi/custom_modules` to `PhysiCell-X/custom_modules`
|
|
|
|
|
|
|
|
4. Copy directory `Physicell-X/sample_projects/pred_prey_mpi/config` to `PhysiCell-X/config`.
|
|
|
|
|
|
|
|
Now, the user can modify `PhysiCell-X/main.cpp`, `PhysiCell-X/Makefile` and the source code files in `PhysiCell-X/custom_modules`.
|
|
|
|
|
|
|
|
Once the user has modified the files (or just wants to run an unmodified example) the project is *built* by typing:
|
|
|
|
|
|
|
|
make
|
|
|
|
|
|
|
|
As an example, when we run `make` *after* checking out the project named `spheroid_tnf_model_mpi`, it produces an executable named `physiboss-tnf-model-mpi`.
|
|
|
|
The name of the executable can be changed by changing the variable named `PROGRAM_NAME` in the `Makefile`.
|
|
|
|
|
|
|
|
The following is a sample output that is produced *after* checking out the `physiboss-tnf-model-mpi` project (`make physiboss-tnf-model-mpi`) and *after* executing `make`.
|
|
|
|
|
|
|
|
python3 beta/setup_libmaboss.py
|
|
|
|
operating system = Linux
|
|
|
|
libMaBoSS will now be installed into the addon PhysiBoSS addon folder:
|
|
|
|
addons/PhysiBoSS
|
|
|
|
|
|
|
|
Beginning download of libMaBoSS into addons/PhysiBoSS ...
|
|
|
|
http://maboss.curie.fr/pub/libMaBoSS-linux64.tar.gz
|
|
|
|
my_file = addons/PhysiBoSS/libMaBoSS-linux64.tar.gz
|
|
|
|
100.1% 2113536 / 2112151
|
|
|
|
installing (uncompressing) the file...
|
|
|
|
Done.
|
|
|
|
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_vector.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_mesh.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_microenvironment.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_solvers.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_matlab.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_utilities.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_basic_agent.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_MultiCellDS.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_agent_container.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/pugixml.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./core/PhysiCell_phenotype.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./core/PhysiCell_cell_container.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./core/PhysiCell_standard_models.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g
|
|
|
|
-DADDON_PHYSIBOSS -I/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/include -DMAXNODES=64 -c ./core/PhysiCell_cell.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./core/PhysiCell_custom.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./core/PhysiCell_utilities.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./core/PhysiCell_constants.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./modules/PhysiCell_SVG.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./modules/PhysiCell_pathology.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./modules/PhysiCell_MultiCellDS.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./modules/PhysiCell_various_outputs.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./modules/PhysiCell_pugixml.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./modules/PhysiCell_settings.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./DistPhy/DistPhy_Environment.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./DistPhy/DistPhy_Cartesian.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./DistPhy/DistPhy_Utils.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./DistPhy/DistPhy_Collective.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g
|
|
|
|
-DADDON_PHYSIBOSS -I/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/include -DMAXNODES=64 -c ./custom_modules/custom.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g
|
|
|
|
-DADDON_PHYSIBOSS -I/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/include -DMAXNODES=64 -c ./custom_modules/submodel_data_structures.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g
|
|
|
|
-DADDON_PHYSIBOSS -I/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/include -DMAXNODES=64 -c ./custom_modules/tnf_receptor_dynamics.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g
|
|
|
|
-DADDON_PHYSIBOSS -I/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/include -DMAXNODES=64 -c ./custom_modules/tnf_boolean_model_interface.cpp
|
|
|
|
compiling
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g
|
|
|
|
-DADDON_PHYSIBOSS -I/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/include -DMAXNODES=64 -c ./addons/PhysiBoSS/src/maboss_network.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g
|
|
|
|
-DADDON_PHYSIBOSS -I/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/include -DMAXNODES=64 -c ./addons/PhysiBoSS/src/maboss_intracellular.cpp
|
|
|
|
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g
|
|
|
|
-DADDON_PHYSIBOSS -I/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/include -DMAXNODES=64
|
|
|
|
-o spheroid_TNF_model_mpi BioFVM_vector.o BioFVM_mesh.o BioFVM_microenvironment.o BioFVM_solvers.o BioFVM_matlab.o
|
|
|
|
BioFVM_utilities.o BioFVM_basic_agent.o BioFVM_MultiCellDS.o BioFVM_agent_container.o pugixml.o PhysiCell_phenotype.o
|
|
|
|
PhysiCell_cell_container.o PhysiCell_standard_models.o PhysiCell_cell.o PhysiCell_custom.o PhysiCell_utilities.o
|
|
|
|
PhysiCell_constants.o PhysiCell_SVG.o PhysiCell_pathology.o PhysiCell_MultiCellDS.o PhysiCell_various_outputs.o
|
|
|
|
PhysiCell_pugixml.o PhysiCell_settings.o DistPhy_Environment.o DistPhy_Cartesian.o DistPhy_Utils.o DistPhy_Collective.o
|
|
|
|
custom.o submodel_data_structures.o tnf_receptor_dynamics.o tnf_boolean_model_interface.o maboss_network.o
|
|
|
|
maboss_intracellular.o main.cpp -L/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/lib -lMaBoSS-static -ldl
|
|
|
|
|
|
|
|
check for spheroid_TNF_model_mpi
|
|
|
|
|
|
|
|
|
|
|
|
An important point to note from the compilation output above is that `libMaBoss` is *downloaded* and installed from the command line.
|
|
|
|
Thus, the machine that this compilation is done on will need *access to the Internet*. This is *not* the case for examples that *do not* need PhysiBoss.
|
|
|
|
By default, everytime when a `make clean` is performed, it *also* deletes the `libMaBoss` library.
|
|
|
|
To prevent this from happening, we need to modify the `Makefile` by *not* calling the `MaBoSS-clean` action when a `make clean` is performed. To be precise, we need to delete the `MaBoSS-clean` from the `clean` rule (see the following code snippet from the `Makefile`).
|
|
|
|
|
|
|
|
clean: MaBoSS-clean #<---Delete this MaBoss-clean
|
|
|
|
Once the user has modified the files (or just wants to run an unmodified example) the project is _built_ by typing:
|
|
|
|
|
|
|
|
```plaintext
|
|
|
|
make
|
|
|
|
```
|
|
|
|
|
|
|
|
As an example, when we run `make` _after_ checking out the project named `spheroid_tnf_model_mpi`, it produces an executable named `physiboss-tnf-model-mpi`. The name of the executable can be changed by changing the variable named `PROGRAM_NAME` in the `Makefile`.
|
|
|
|
|
|
|
|
The following is a sample output that is produced _after_ checking out the `physiboss-tnf-model-mpi` project (`make physiboss-tnf-model-mpi`) and _after_ executing `make`.
|
|
|
|
|
|
|
|
```plaintext
|
|
|
|
python3 beta/setup_libmaboss.py
|
|
|
|
operating system = Linux
|
|
|
|
libMaBoSS will now be installed into the addon PhysiBoSS addon folder:
|
|
|
|
addons/PhysiBoSS
|
|
|
|
|
|
|
|
Beginning download of libMaBoSS into addons/PhysiBoSS ...
|
|
|
|
http://maboss.curie.fr/pub/libMaBoSS-linux64.tar.gz
|
|
|
|
my_file = addons/PhysiBoSS/libMaBoSS-linux64.tar.gz
|
|
|
|
100.1% 2113536 / 2112151
|
|
|
|
installing (uncompressing) the file...
|
|
|
|
Done.
|
|
|
|
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_vector.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_mesh.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_microenvironment.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_solvers.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_matlab.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_utilities.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_basic_agent.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_MultiCellDS.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/BioFVM_agent_container.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./BioFVM/pugixml.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./core/PhysiCell_phenotype.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./core/PhysiCell_cell_container.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./core/PhysiCell_standard_models.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g
|
|
|
|
-DADDON_PHYSIBOSS -I/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/include -DMAXNODES=64 -c ./core/PhysiCell_cell.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./core/PhysiCell_custom.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./core/PhysiCell_utilities.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./core/PhysiCell_constants.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./modules/PhysiCell_SVG.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./modules/PhysiCell_pathology.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./modules/PhysiCell_MultiCellDS.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./modules/PhysiCell_various_outputs.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./modules/PhysiCell_pugixml.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./modules/PhysiCell_settings.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./DistPhy/DistPhy_Environment.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./DistPhy/DistPhy_Cartesian.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./DistPhy/DistPhy_Utils.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g -c ./DistPhy/DistPhy_Collective.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g
|
|
|
|
-DADDON_PHYSIBOSS -I/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/include -DMAXNODES=64 -c ./custom_modules/custom.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g
|
|
|
|
-DADDON_PHYSIBOSS -I/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/include -DMAXNODES=64 -c ./custom_modules/submodel_data_structures.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g
|
|
|
|
-DADDON_PHYSIBOSS -I/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/include -DMAXNODES=64 -c ./custom_modules/tnf_receptor_dynamics.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g
|
|
|
|
-DADDON_PHYSIBOSS -I/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/include -DMAXNODES=64 -c ./custom_modules/tnf_boolean_model_interface.cpp
|
|
|
|
compiling
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g
|
|
|
|
-DADDON_PHYSIBOSS -I/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/include -DMAXNODES=64 -c ./addons/PhysiBoSS/src/maboss_network.cpp
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g
|
|
|
|
-DADDON_PHYSIBOSS -I/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/include -DMAXNODES=64 -c ./addons/PhysiBoSS/src/maboss_intracellular.cpp
|
|
|
|
|
|
|
|
mpic++ -march=native -O3 -fomit-frame-pointer -mfpmath=both -fopenmp -m64 -std=c++11 -g
|
|
|
|
-DADDON_PHYSIBOSS -I/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/include -DMAXNODES=64
|
|
|
|
-o spheroid_TNF_model_mpi BioFVM_vector.o BioFVM_mesh.o BioFVM_microenvironment.o BioFVM_solvers.o BioFVM_matlab.o
|
|
|
|
BioFVM_utilities.o BioFVM_basic_agent.o BioFVM_MultiCellDS.o BioFVM_agent_container.o pugixml.o PhysiCell_phenotype.o
|
|
|
|
PhysiCell_cell_container.o PhysiCell_standard_models.o PhysiCell_cell.o PhysiCell_custom.o PhysiCell_utilities.o
|
|
|
|
PhysiCell_constants.o PhysiCell_SVG.o PhysiCell_pathology.o PhysiCell_MultiCellDS.o PhysiCell_various_outputs.o
|
|
|
|
PhysiCell_pugixml.o PhysiCell_settings.o DistPhy_Environment.o DistPhy_Cartesian.o DistPhy_Utils.o DistPhy_Collective.o
|
|
|
|
custom.o submodel_data_structures.o tnf_receptor_dynamics.o tnf_boolean_model_interface.o maboss_network.o
|
|
|
|
maboss_intracellular.o main.cpp -L/gpfs/home/bsc99/bsc99102/GS_PhysiCell_X/addons/PhysiBoSS/MaBoSS-env-2.0/engine/lib -lMaBoSS-static -ldl
|
|
|
|
|
|
|
|
check for spheroid_TNF_model_mpi
|
|
|
|
```
|
|
|
|
|
|
|
|
An important point to note from the compilation output above is that `libMaBoss` is _downloaded_ and installed from the command line. Thus, the machine that this compilation is done on will need _access to the Internet_. This is _not_ the case for examples that _do not_ need PhysiBoss. By default, everytime when a `make clean` is performed, it _also_ deletes the `libMaBoss` library. To prevent this from happening, we need to modify the `Makefile` by _not_ calling the `MaBoSS-clean` action when a `make clean` is performed. To be precise, we need to delete the `MaBoSS-clean` from the `clean` rule (see the following code snippet from the `Makefile`).
|
|
|
|
|
|
|
|
```plaintext
|
|
|
|
clean: MaBoSS-clean #<---Delete this MaBoss-clean
|
|
|
|
rm -f *.o
|
|
|
|
rm -f $(PROGRAM_NAME)*
|
|
|
|
```
|
|
|
|
|
|
|
|
> **📝 IMPORTANT: Note on libMaBoss library**
|
|
|
|
|
|
|
|
> It is only downloaded the very first time but subsequently its deletion & download can be prevented by modifying the `clean` rule in the `Makefile` as shown above.
|
|
|
|
|
|
|
|
To run the executable produced above i.e. `spheroid_TNF_model_mpi` above in parallel (MPI + OpenMP) on an HPC system/cluster, we *typically* use a submission script written in the Unix shell script language.
|
|
|
|
This does *not* mean that the program cannot run on a Laptop, Desktop etc. but the syntax for execution varies (and most certainly the performance gains are smaller).
|
|
|
|
We show below a submission script for the SLURM workload manager for HPC systems and explain every line of the script.
|
|
|
|
```bash
|
|
|
|
To run the executable produced above i.e. `spheroid_TNF_model_mpi` above in parallel (MPI + OpenMP) on an HPC system/cluster, we _typically_ use a submission script written in the Unix shell script language. This does _not_ mean that the program cannot run on a Laptop, Desktop etc. but the syntax for execution varies (and most certainly the performance gains are smaller). We show below a submission script for the SLURM workload manager for HPC systems and explain every line of the script.
|
|
|
|
|
|
|
|
```shell
|
|
|
|
#!/bin/bash
|
|
|
|
|
|
|
|
#SBATCH --job-name="TNF_Simulation"
|
| ... | ... | @@ -356,47 +273,28 @@ We show below a submission script for the SLURM workload manager for HPC systems |
|
|
|
|
|
|
|
mpiexec ./spheroid_TNF_model_mpi
|
|
|
|
```
|
|
|
|
We assume that the script above is saved as `script_tnf_mpi.sh`.
|
|
|
|
The following describes (almost) every line:
|
|
|
|
|
|
|
|
1. Line 1 means that this script is to be executed using the Linux `bash` shell.
|
|
|
|
|
|
|
|
2. Line 2 gives a name to this job - in this case "TNF\_simulation".
|
|
|
|
|
|
|
|
3. Line 3 means assign 150 HPC nodes to this job. (In our experiments we use HPC nodes which have 48 cores. These 48 cores are distributed as 2 sockets of 24 cores each.)
|
|
|
|
|
|
|
|
4. Line 4 states that we only need 1 MPI process per node. This means we will only have 150 MPI processes (= the number of HPC nodes requested).
|
|
|
|
|
|
|
|
5. Line 5 says that 48 OpenMP threads should be spawned per MPI process. Each of these threads will "cling" (or technically bind) to a core (remember we have 48 cores and 48 threads). Thus, the total number of cores being used in this job are: $150 \times 48 = 7200$ i.e. 150 MPI processes times 48 OpenMP threads.
|
|
|
|
|
|
|
|
6. Line 6 means that this job can execute for a maximum of 72 hours.
|
|
|
|
|
|
|
|
7. Line 7 says that the output of the job should be written to a file named `output-[jobid]`, where `jobid` is a unique number that is provided by SLURM.
|
|
|
|
|
|
|
|
8. Line 8 says exactly the same thing as Line 7 but about the error file.
|
|
|
|
|
|
|
|
9. Line 9 requests "exclusive" nodes for our job i.e. no other user's job can be run in the nodes allocated to our job while our job is running. This is very important for performance.
|
|
|
|
|
|
|
|
We assume that the script above is saved as `script_tnf_mpi.sh`. The following describes (almost) every line:
|
|
|
|
|
|
|
|
1. Line 1 means that this script is to be executed using the Linux `bash` shell.
|
|
|
|
2. Line 2 gives a name to this job - in this case "TNF_simulation".
|
|
|
|
3. Line 3 means assign 150 HPC nodes to this job. (In our experiments we use HPC nodes which have 48 cores. These 48 cores are distributed as 2 sockets of 24 cores each.)
|
|
|
|
4. Line 4 states that we only need 1 MPI process per node. This means we will only have 150 MPI processes (= the number of HPC nodes requested).
|
|
|
|
5. Line 5 says that 48 OpenMP threads should be spawned per MPI process. Each of these threads will "cling" (or technically bind) to a core (remember we have 48 cores and 48 threads). Thus, the total number of cores being used in this job are: $150 \\times 48 = 7200$ i.e. 150 MPI processes times 48 OpenMP threads.
|
|
|
|
6. Line 6 means that this job can execute for a maximum of 72 hours.
|
|
|
|
7. Line 7 says that the output of the job should be written to a file named `output-[jobid]`, where `jobid` is a unique number that is provided by SLURM.
|
|
|
|
8. Line 8 says exactly the same thing as Line 7 but about the error file.
|
|
|
|
9. Line 9 requests "exclusive" nodes for our job i.e. no other user's job can be run in the nodes allocated to our job while our job is running. This is very important for performance.
|
|
|
|
10. Line 10 directs the OpenMP thread binding details and environment variable values to be printed in the error file.
|
|
|
|
|
|
|
|
11. Line 11 fixes the "static" schedule for the OpenMP parallel `for` loops (when they are executed).
|
|
|
|
|
|
|
|
12. Line 12 says that the number of OpenMP threads which are spawned should be equal to the value of the SLURM variable `SLURM_CPUS_PER_TASK`, which in this case is 48.
|
|
|
|
|
|
|
|
13. Line 13 tells the OpenMP environment that threads should be placed as far apart from each other as possible. In this case it does not matter as we have 48 threads and 48 cores i.e. the whole node is *completely* filled with one thread per core.
|
|
|
|
|
|
|
|
13. Line 13 tells the OpenMP environment that threads should be placed as far apart from each other as possible. In this case it does not matter as we have 48 threads and 48 cores i.e. the whole node is _completely_ filled with one thread per core.
|
|
|
|
14. Line 14 tells the OpenMP threads to bind to "threads". Our HPC node runs only one thread per core (no hyper-threading) and hence the option "threads" in this case translates to "cores".
|
|
|
|
|
|
|
|
15. Line 15 finally executes the program. through `mpiexec` - the MPI launch command.
|
|
|
|
|
|
|
|
The script above *may* not fetch us the best performance.
|
|
|
|
In HPC, the software *must* map to the underlying hardware in order to extract maximal performance.
|
|
|
|
Thus, to continue this discussion, we present another script next that gives better performance on our systems.
|
|
|
|
The reader may recollect that our experiments are performed on HPC nodes having 48 cores each and organized as 2 sockets of 24 cores each.
|
|
|
|
Our aim is to spawn 1 MPI process per socket and 24 threads per MPI process to bind to the 24 cores in each socket.
|
|
|
|
This reduces *false sharing* between OpenMP threads - a topic which is beyond the scope of the document but can be found easily by consulting any book on OpenMP.
|
|
|
|
The script is very similar to the script presented previously and is shown below:
|
|
|
|
```bash
|
|
|
|
The script above _may_ not fetch us the best performance. In HPC, the software _must_ map to the underlying hardware in order to extract maximal performance. Thus, to continue this discussion, we present another script next that gives better performance on our systems. The reader may recollect that our experiments are performed on HPC nodes having 48 cores each and organized as 2 sockets of 24 cores each. Our aim is to spawn 1 MPI process per socket and 24 threads per MPI process to bind to the 24 cores in each socket. This reduces _false sharing_ between OpenMP threads - a topic which is beyond the scope of the document but can be found easily by consulting any book on OpenMP. The script is very similar to the script presented previously and is shown below:
|
|
|
|
|
|
|
|
```shell
|
|
|
|
#!/bin/bash
|
|
|
|
|
|
|
|
#SBATCH --job-name="TNF_Simulation"
|
| ... | ... | @@ -417,30 +315,25 @@ The script is very similar to the script presented previously and is shown below |
|
|
|
mpiexec --map-by ppr:1:socket:pe=24 ./spheroid_TNF_model_mpi
|
|
|
|
```
|
|
|
|
|
|
|
|
The difference in this script from the previous script is that we are creating 2 MPI processes per node (or 1 MPI process per socket and we have 2 sockets in one node).
|
|
|
|
Further, we are now spawning 24 OpenMP threads per MPI process. The *total* number of threads in this script and the previous script are exactly the same i.e. 7200 threads (with one thread per core).
|
|
|
|
Thus, the total number of cores that we use in both the scripts are 7200.
|
|
|
|
The second script maps the MPI processes more properly to the internal architecture of the node as the node consist of 2 sockets.
|
|
|
|
The other difference is the `–map-by ppr` syntax. `ppr` stands for processes per resource and here we have 1 MPI process per socket (indicated by the `:socket` in the `mpiexec` statement).
|
|
|
|
Further, the number of *processing elements* (given by `pe` in the `mpiexec` statement) are 24 in each *resource*.
|
|
|
|
The processing elements are equivalent to our cores.
|
|
|
|
Thus, in a nutshell the `mpiexec` statement indicates the hardware resources i.e. sockets and cores and the overall script maps the MPI processes to sockets and the OpenMP threads to cores.
|
|
|
|
The user is encouraged to experiment with both the scripts (or modify the values of the variables in the script to cater to his/her machine's architecture) and to observe the difference in performance.
|
|
|
|
The difference in this script from the previous script is that we are creating 2 MPI processes per node (or 1 MPI process per socket and we have 2 sockets in one node). Further, we are now spawning 24 OpenMP threads per MPI process. The _total_ number of threads in this script and the previous script are exactly the same i.e. 7200 threads (with one thread per core). Thus, the total number of cores that we use in both the scripts are 7200. The second script maps the MPI processes more properly to the internal architecture of the node as the node consist of 2 sockets. The other difference is the `–map-by ppr` syntax. `ppr` stands for processes per resource and here we have 1 MPI process per socket (indicated by the `:socket` in the `mpiexec` statement). Further, the number of _processing elements_ (given by `pe` in the `mpiexec` statement) are 24 in each _resource_. The processing elements are equivalent to our cores. Thus, in a nutshell the `mpiexec` statement indicates the hardware resources i.e. sockets and cores and the overall script maps the MPI processes to sockets and the OpenMP threads to cores. The user is encouraged to experiment with both the scripts (or modify the values of the variables in the script to cater to his/her machine's architecture) and to observe the difference in performance.
|
|
|
|
|
|
|
|
## Running the TNF MPI example
|
|
|
|
|
|
|
|
We now detail out how to run the TNF MPI example. We assume that the
|
|
|
|
project is in a clean state i.e. PhysiCell-X is just downloaded/cloned.
|
|
|
|
The first step is to see the projects which are available using
|
|
|
|
We now detail out how to run the TNF MPI example. We assume that the project is in a clean state i.e. PhysiCell-X is just downloaded/cloned. The first step is to see the projects which are available using
|
|
|
|
|
|
|
|
make list-projects
|
|
|
|
```plaintext
|
|
|
|
make list-projects
|
|
|
|
```
|
|
|
|
|
|
|
|
. We must now choose to check-out the TNF MPI project using
|
|
|
|
|
|
|
|
make physiboss-tnf-model-mpi
|
|
|
|
```plaintext
|
|
|
|
make physiboss-tnf-model-mpi
|
|
|
|
```
|
|
|
|
|
|
|
|
This performs the following actions (as explained above):
|
|
|
|
```bash
|
|
|
|
|
|
|
|
```shell
|
|
|
|
cp ./sample_projects_intracellular/boolean/spheroid_tnf_model_mpi/custom_modules/*
|
|
|
|
./custom_modules/
|
|
|
|
touch main.cpp && cp main.cpp main-backup.cpp
|
| ... | ... | @@ -451,38 +344,27 @@ This performs the following actions (as explained above): |
|
|
|
cp ./config/PhysiCell_settings.xml ./config/PhysiCell_settings-backup.xml
|
|
|
|
cp ./sample_projects_intracellular/boolean/spheroid_tnf_model_mpi/config/* ./config/
|
|
|
|
```
|
|
|
|
At this point please make sure that you have a C++ compiler (like GCC)
|
|
|
|
and MPI implementation (like OpenMPI) configured properly. We next
|
|
|
|
compile this checked-out project by simply executing
|
|
|
|
|
|
|
|
make
|
|
|
|
|
|
|
|
. If the compilation is successful, users should see an executable named
|
|
|
|
`spheroid_TNF_model_mpi`. There are several submission script files
|
|
|
|
provided in the top level directory and in this example we use the
|
|
|
|
submission script named `script_physiboss_tnf_model_mpi.sh`. The user is
|
|
|
|
encouraged to open this file using a text editor and understand the
|
|
|
|
parameters (as explained in the section above) so that even if a job
|
|
|
|
manager like SLURM or PBS is not available, he/she is able to translate
|
|
|
|
the parameters to execute it on the local system. If the SLURM system is
|
|
|
|
available (like on the Marenostrum supercomputer), this job can simply
|
|
|
|
be submitted (and subsequently executed) using
|
|
|
|
|
|
|
|
sbatch script_physiboss_tnf_model_mpi.sh
|
|
|
|
|
|
|
|
. However before submitting this job for running, it is better to check
|
|
|
|
and understand some parameters. We start with the submission script
|
|
|
|
itself and explain sequence which should be followed to perform a basic
|
|
|
|
quick check of the requirements. In the submisssion script, the line
|
|
|
|
```bash
|
|
|
|
|
|
|
|
At this point please make sure that you have a C++ compiler (like GCC) and MPI implementation (like OpenMPI) configured properly. We next compile this checked-out project by simply executing
|
|
|
|
|
|
|
|
```plaintext
|
|
|
|
make
|
|
|
|
```
|
|
|
|
|
|
|
|
. If the compilation is successful, users should see an executable named `spheroid_TNF_model_mpi`. There are several submission script files provided in the top level directory and in this example we use the submission script named `script_physiboss_tnf_model_mpi.sh`. The user is encouraged to open this file using a text editor and understand the parameters (as explained in the section above) so that even if a job manager like SLURM or PBS is not available, he/she is able to translate the parameters to execute it on the local system. If the SLURM system is available (like on the Marenostrum supercomputer), this job can simply be submitted (and subsequently executed) using
|
|
|
|
|
|
|
|
```plaintext
|
|
|
|
sbatch script_physiboss_tnf_model_mpi.sh
|
|
|
|
```
|
|
|
|
|
|
|
|
. However before submitting this job for running, it is better to check and understand some parameters. We start with the submission script itself and explain sequence which should be followed to perform a basic quick check of the requirements. In the submisssion script, the line
|
|
|
|
|
|
|
|
```shell
|
|
|
|
mpiexec --map-by ppr:1:socket:pe=24 --report-bindings ./spheroid_TNF_model_mpi
|
|
|
|
```
|
|
|
|
indicates that we do *not* input any special settings file to the
|
|
|
|
executable program `spheroid_TNF_model_mpi`. If you want to input any
|
|
|
|
special settings file, say `PhysiCell_settings_my_file.xml` then this
|
|
|
|
must be given after the name of the executable. Thus, we are using the
|
|
|
|
default `PhysiCell_settings.xml` file in the `config` directory. If we
|
|
|
|
open the `Physicell_settings.xml` file, we see the domain dimensions in the bery beginning of the file as:
|
|
|
|
|
|
|
|
indicates that we do _not_ input any special settings file to the executable program `spheroid_TNF_model_mpi`. If you want to input any special settings file, say `PhysiCell_settings_my_file.xml` then this must be given after the name of the executable. Thus, we are using the default `PhysiCell_settings.xml` file in the `config` directory. If we open the `Physicell_settings.xml` file, we see the domain dimensions in the bery beginning of the file as:
|
|
|
|
|
|
|
|
```xml
|
|
|
|
<domain>
|
|
|
|
<x_min>-200</x_min>
|
| ... | ... | @@ -498,82 +380,54 @@ open the `Physicell_settings.xml` file, we see the domain dimensions in the bery |
|
|
|
</domain>
|
|
|
|
```
|
|
|
|
|
|
|
|
First, this shows that domain dimensions in the X/Y and Z direction vary
|
|
|
|
from $[-200,+200]$ i.e. $400$ units of length. Second, the length/width
|
|
|
|
and height of the *diffusion* voxel is $20$. Please note that the
|
|
|
|
length/breadth/height of the *mechanical* voxel is specified through the
|
|
|
|
`main.cpp` of the specific project. Further, since PhysiCell-X works
|
|
|
|
*only* for 3-D problems, the 2-D settings are set to `false`. The total
|
|
|
|
number of diffusion voxels in this case in the X, Y and Z directions are
|
|
|
|
$\frac{200-(-200)}{20} = \frac{400}{20} = 20$ each. If we check in the
|
|
|
|
`script_physiboss_tnf_model_mpi.sh` file, we can see that the total
|
|
|
|
number of MPI processes are only *two* i.e. we have a single node with 2
|
|
|
|
MPI processes per node as indicated by the lines below:
|
|
|
|
```bash
|
|
|
|
First, this shows that domain dimensions in the X/Y and Z direction vary from $\[-200,+200\]$ i.e. $400$ units of length. Second, the length/width and height of the _diffusion_ voxel is $20$. Please note that the length/breadth/height of the _mechanical_ voxel is specified through the `main.cpp` of the specific project. Further, since PhysiCell-X works _only_ for 3-D problems, the 2-D settings are set to `false`. The total number of diffusion voxels in this case in the X, Y and Z directions are $\\frac{200-(-200)}{20} = \\frac{400}{20} = 20$ each. If we check in the `script_physiboss_tnf_model_mpi.sh` file, we can see that the total number of MPI processes are only _two_ i.e. we have a single node with 2 MPI processes per node as indicated by the lines below:
|
|
|
|
|
|
|
|
```shell
|
|
|
|
#SBATCH --nodes=1
|
|
|
|
#SBATCH --ntasks-per-node=2
|
|
|
|
```
|
|
|
|
|
|
|
|
One of the conditions that must be fulfilled is that the total number of
|
|
|
|
diffusion (or mechanical) voxels in the X-direction *must* be
|
|
|
|
*completely/exactly/perfectly* divisible by the total number of MPI
|
|
|
|
processes. In this case, we have a total of 20 diffusion voxels and 2
|
|
|
|
MPI processes and we can see that $\frac{20}{2} = 10$ gives 10
|
|
|
|
*diffusion* voxels per MPI process. This divisibility condition is not
|
|
|
|
needed in the Y/Z directions as we only implement a 1-dimensional
|
|
|
|
X-direction decomposition. At this stage it is very important to check
|
|
|
|
the size of the mechanical voxel in the `main.cpp` file in the top level
|
|
|
|
directory and the following line in this file shows that the size of the
|
|
|
|
mechanical voxel is also set to 20 (i.e. the same as the size of the
|
|
|
|
diffusion voxel):
|
|
|
|
One of the conditions that must be fulfilled is that the total number of diffusion (or mechanical) voxels in the X-direction _must_ be _completely/exactly/perfectly_ divisible by the total number of MPI processes. In this case, we have a total of 20 diffusion voxels and 2 MPI processes and we can see that $\\frac{20}{2} = 10$ gives 10 _diffusion_ voxels per MPI process. This divisibility condition is not needed in the Y/Z directions as we only implement a 1-dimensional X-direction decomposition. At this stage it is very important to check the size of the mechanical voxel in the `main.cpp` file in the top level directory and the following line in this file shows that the size of the mechanical voxel is also set to 20 (i.e. the same as the size of the diffusion voxel):
|
|
|
|
|
|
|
|
double mechanics_voxel_size = 20;
|
|
|
|
|
|
|
|
It is clear that the total number of mechanical voxels in the X-direction i.e. $\frac{200-(-200)}{20}=20$ are also divisible by the
|
|
|
|
total number of MPI processes (2 in this case). Thus, we have checked
|
|
|
|
that
|
|
|
|
```plaintext
|
|
|
|
double mechanics_voxel_size = 20;
|
|
|
|
```
|
|
|
|
|
|
|
|
1. Mechanical voxel size $\geq$ Diffusion voxel size.
|
|
|
|
It is clear that the total number of mechanical voxels in the X-direction i.e. $\\frac{200-(-200)}{20}=20$ are also divisible by the total number of MPI processes (2 in this case). Thus, we have checked that
|
|
|
|
|
|
|
|
1. Mechanical voxel size $\\geq$ Diffusion voxel size.
|
|
|
|
2. Total number of voxels (both Mechanical and Diffusion) in the X-direction are perfectly divisible by the total number of MPI processes.
|
|
|
|
|
|
|
|
The `PhysiCell_settings.xml` file also indicates that the total number
|
|
|
|
of OpenMP threads is just one i.e.
|
|
|
|
The `PhysiCell_settings.xml` file also indicates that the total number of OpenMP threads is just one i.e.
|
|
|
|
|
|
|
|
```xml
|
|
|
|
<parallel>
|
|
|
|
<omp_num_threads>1</omp_num_threads>
|
|
|
|
</parallel>
|
|
|
|
```
|
|
|
|
This has no effect on our program *because* we have commented out the
|
|
|
|
line in `main.cpp` that sets the number of OpenMP threads to the value
|
|
|
|
above i.e. just one OpenMP thread. See the line below in `main.cpp`.
|
|
|
|
```bash
|
|
|
|
|
|
|
|
This has no effect on our program _because_ we have commented out the line in `main.cpp` that sets the number of OpenMP threads to the value above i.e. just one OpenMP thread. See the line below in `main.cpp`.
|
|
|
|
|
|
|
|
```shell
|
|
|
|
//omp_set_num_threads(PhysiCell_settings.omp_num_threads); <--- We use OMP_NUM_THREADS
|
|
|
|
```
|
|
|
|
Instead of setting the number of OpenMP threads using this XML
|
|
|
|
parameter, we use the `OMP_NUM_THREADS` environment variable as given in
|
|
|
|
`script_physiboss_tnf_model_mpi.sh`. In this file, we simply set this
|
|
|
|
environment variable as shown below:
|
|
|
|
```bash
|
|
|
|
|
|
|
|
Instead of setting the number of OpenMP threads using this XML parameter, we use the `OMP_NUM_THREADS` environment variable as given in `script_physiboss_tnf_model_mpi.sh`. In this file, we simply set this environment variable as shown below:
|
|
|
|
|
|
|
|
```shell
|
|
|
|
export OMP_NUM_THREADS=$SLURM_CPUS_PER_TASK
|
|
|
|
```
|
|
|
|
|
|
|
|
where the environment variable `SLURM_CPUS_PER_TASK` indicate the number
|
|
|
|
of OpenMP threads (SLURM terminology is not intuitive !). This variable
|
|
|
|
is set as (see `script_physiboss_tnf_model_mpi.sh`):
|
|
|
|
```bash
|
|
|
|
where the environment variable `SLURM_CPUS_PER_TASK` indicate the number of OpenMP threads (SLURM terminology is not intuitive !). This variable is set as (see `script_physiboss_tnf_model_mpi.sh`):
|
|
|
|
|
|
|
|
```shell
|
|
|
|
#SBATCH --cpus-per-task=24
|
|
|
|
```
|
|
|
|
Here, the environment variable `SLURM_CPUS_PER_TASK` takes its value
|
|
|
|
from the `–cpus-per-task=24` SLURM option. Thus, the number of OpenMP
|
|
|
|
threads in our program is 24 per socket (and $2 \times 24 = 48$ per
|
|
|
|
node).\
|
|
|
|
The initial part of the `main.cpp` contains the lines of code to build a
|
|
|
|
Cartesian topology. It can be noted that *this part of the code will
|
|
|
|
remain exactly the same for all the parallel programs*. Thus, the user
|
|
|
|
can simply copy and paste this code into any new programs that they
|
|
|
|
make. We show this part of the code below:
|
|
|
|
```
|
|
|
|
|
|
|
|
Here, the environment variable `SLURM_CPUS_PER_TASK` takes its value from the `–cpus-per-task=24` SLURM option. Thus, the number of OpenMP threads in our program is 24 per socket (and $2 \\times 24 = 48$ per node).\
|
|
|
|
The initial part of the `main.cpp` contains the lines of code to build a Cartesian topology. It can be noted that _this part of the code will remain exactly the same for all the parallel programs_. Thus, the user can simply copy and paste this code into any new programs that they make. We show this part of the code below:
|
|
|
|
|
|
|
|
```plaintext
|
|
|
|
/*=======================================================================================*/
|
|
|
|
/* Create mpi_Environment object, initialize it, then create Cartesian Topology */
|
|
|
|
/*=======================================================================================*/
|
| ... | ... | @@ -592,24 +446,16 @@ make. We show this part of the code below: |
|
|
|
return 0;
|
|
|
|
```
|
|
|
|
|
|
|
|
If you not familiar with parallel programming and this appears strange
|
|
|
|
to you, then we strongly suggest *not* devoting time to understanding
|
|
|
|
this piece of the code as this code is repeated as it for *all* the
|
|
|
|
programs. However, we encourage the user to check out the `DistPhy`
|
|
|
|
directory and the `.h` files under it to get an idea of what data
|
|
|
|
members and functions the classes `mpi_Environment` and `mpi_Cartesian`
|
|
|
|
contain. We could as developers have abstracted away some of the calls
|
|
|
|
above but we prefer to let the user know (to an extent) the parallel
|
|
|
|
calls that are being executed behind the scenes.\
|
|
|
|
If you not familiar with parallel programming and this appears strange to you, then we strongly suggest _not_ devoting time to understanding this piece of the code as this code is repeated as it for _all_ the programs. However, we encourage the user to check out the `DistPhy` directory and the `.h` files under it to get an idea of what data members and functions the classes `mpi_Environment` and `mpi_Cartesian` contain. We could as developers have abstracted away some of the calls above but we prefer to let the user know (to an extent) the parallel calls that are being executed behind the scenes.\
|
|
|
|
After compilation using `make`, we can now submit the code using
|
|
|
|
|
|
|
|
```shell
|
|
|
|
sbatch script_physiboss_tnf_model_mpi.sh
|
|
|
|
```
|
|
|
|
|
|
|
|
. If everything goes well (fingers crossed !), we should see an output
|
|
|
|
like:
|
|
|
|
```
|
|
|
|
. If everything goes well (fingers crossed !), we should see an output like:
|
|
|
|
|
|
|
|
```plaintext
|
|
|
|
|
|
|
|
____ _ _ _____ _ _ __ __ _ ___ __
|
|
|
|
| __ \| | (_)/ ____| | | | \ \ / / (_) / _ \/_ |
|
| ... | ... | @@ -736,79 +582,47 @@ Total simulation runtime: |
|
|
|
0 days, 0 hours, 0 minutes, and 49.0327 seconds
|
|
|
|
```
|
|
|
|
|
|
|
|
In addition to giving the various input files such as
|
|
|
|
`PhysiCell_settings.xml, config_5k.txt`, it also gives the cells/agents
|
|
|
|
on each MPI process (initially).
|
|
|
|
|
|
|
|
For example, the number of cells on MPI
|
|
|
|
process Rank = 1 are 2500 and that on MPI process Rank = 0 are 2501.
|
|
|
|
This information can be useful as it gives us an idea of the work load
|
|
|
|
on each process (ideally we would want this to be as balanced as
|
|
|
|
possible). After the initial time, it is *only* the total number of
|
|
|
|
cells in the domain that are displayed (and not on individual
|
|
|
|
processes). In this example, the agents or cells first increase, then
|
|
|
|
decrease due to the effect of TNF and then increases again (an
|
|
|
|
alternating increasing/decreasing trend is expected here).
|
|
|
|
In addition to giving the various input files such as `PhysiCell_settings.xml, config_5k.txt`, it also gives the cells/agents on each MPI process (initially).
|
|
|
|
|
|
|
|
Finally, the
|
|
|
|
total simulation time is printed. In addition to this output, there are
|
|
|
|
multiple `.svg, .xml` and `.mat` files written in the `output` directory
|
|
|
|
which can be examined with a browser, text editor or other
|
|
|
|
post-processing applications.
|
|
|
|
For example, the number of cells on MPI process Rank = 1 are 2500 and that on MPI process Rank = 0 are 2501. This information can be useful as it gives us an idea of the work load on each process (ideally we would want this to be as balanced as possible). After the initial time, it is _only_ the total number of cells in the domain that are displayed (and not on individual processes). In this example, the agents or cells first increase, then decrease due to the effect of TNF and then increases again (an alternating increasing/decreasing trend is expected here).
|
|
|
|
|
|
|
|
Finally, the total simulation time is printed. In addition to this output, there are multiple `.svg, .xml` and `.mat` files written in the `output` directory which can be examined with a browser, text editor or other post-processing applications.
|
|
|
|
|
|
|
|
# Future Work
|
|
|
|
|
|
|
|
PhysiCell-X is project that is being actively developed as the distributed-parallel version of PhysiCell \[2\]. Many features are being planned for the future releases of PhysiCell-X which may include:
|
|
|
|
|
|
|
|
PhysiCell-X is project that is being actively developed as the distributed-parallel version of PhysiCell [2].
|
|
|
|
Many features are being planned for the future releases of PhysiCell-X which
|
|
|
|
may include:
|
|
|
|
|
|
|
|
- A generalized, flexible function to set the initial cell positions and an arbitrary number of parameters related to the cells. The input file will be read by *all* the processes to set the cell positions and any other desired parameter.
|
|
|
|
|
|
|
|
- A function to extract the list of neighboring cells of a cell even if they are in the neighboring sub-domain.
|
|
|
|
|
|
|
|
- A facility where a limited number of cells (minimum two) can attach to each other.
|
|
|
|
|
|
|
|
- A completely distributed parallel version of the Thomas algorithm to enhance scalability.
|
|
|
|
|
|
|
|
- A 3-D domain partitioning scheme to incorporate more cores.
|
|
|
|
|
|
|
|
- GPU support for the most computationally intensive kernels of the application.
|
|
|
|
|
|
|
|
- An in-homogeneous domain partitioning scheme to improve the load balance of cells for spherical domains.
|
|
|
|
|
|
|
|
- Multiple parallelized versions of the same function for flexibility.
|
|
|
|
* A generalized, flexible function to set the initial cell positions and an arbitrary number of parameters related to the cells. The input file will be read by _all_ the processes to set the cell positions and any other desired parameter.
|
|
|
|
* A function to extract the list of neighboring cells of a cell even if they are in the neighboring sub-domain.
|
|
|
|
* A facility where a limited number of cells (minimum two) can attach to each other.
|
|
|
|
* A completely distributed parallel version of the Thomas algorithm to enhance scalability.
|
|
|
|
* A 3-D domain partitioning scheme to incorporate more cores.
|
|
|
|
* GPU support for the most computationally intensive kernels of the application.
|
|
|
|
* An in-homogeneous domain partitioning scheme to improve the load balance of cells for spherical domains.
|
|
|
|
* Multiple parallelized versions of the same function for flexibility.
|
|
|
|
|
|
|
|
# Acknowledgements
|
|
|
|
|
|
|
|
- We profusely thank `Vincent Noel` for providing the MPI code for interfacing PhysiBoss, locating memory issues, and sitting through some very painful troubleshooting sessions.
|
|
|
|
|
|
|
|
- Many thanks to `Randy Heiland` and `Paul Macklin` for very patiently answering several questions from the developers and carrying out infinitely interesting discussions (even on Friday evenings !).
|
|
|
|
|
|
|
|
- We are very grateful to `Jose Carbonell` and `Thalia Ntiniakou` for the constant support, correctness verification using PCA and very useful overall advice in our regular meetings.
|
|
|
|
* We profusely thank `Vincent Noel` for providing the MPI code for interfacing PhysiBoss, locating memory issues, and sitting through some very painful troubleshooting sessions.
|
|
|
|
* Many thanks to `Randy Heiland` and `Paul Macklin` for very patiently answering several questions from the developers and carrying out infinitely interesting discussions (even on Friday evenings !).
|
|
|
|
* We are very grateful to `Jose Carbonell` and `Thalia Ntiniakou` for the constant support, correctness verification using PCA and very useful overall advice in our regular meetings.
|
|
|
|
* `Marc Clasca Ramirez` and `Marta Garcia Gasulla` gave extremely useful insights into the performance of the OpenMP version of PhysiCell.
|
|
|
|
* `Shardool Kulkarni` must be thanked for mathematically motivating and guiding us to consider alternatives to the serial Thomas algorithm and (most importantly) trusting us with his final year MSc project !
|
|
|
|
|
|
|
|
- `Marc Clasca Ramirez` and `Marta Garcia Gasulla` gave extremely useful insights into the performance of the OpenMP version of PhysiCell.
|
|
|
|
|
|
|
|
- `Shardool Kulkarni` must be thanked for mathematically motivating and guiding us to consider alternatives to the serial Thomas algorithm and (most importantly) trusting us with his final year MSc project !
|
|
|
|
|
|
|
|
The research leading to these results has received funding from EU H2020
|
|
|
|
Programme under the PerMedCoE project, grant agreement number 951773 and
|
|
|
|
the INFORE project, grant agreement number 825070.
|
|
|
|
The research leading to these results has received funding from EU H2020 Programme under the PerMedCoE project, grant agreement number 951773 and the INFORE project, grant agreement number 825070.
|
|
|
|
|
|
|
|
## Footnotes:
|
|
|
|
|
|
|
|
$^1$: We encourage you to read this tutorial as it will *also* help you understand how to run examples in PhysiCell-X
|
|
|
|
$^1$: We encourage you to read this tutorial as it will _also_ help you understand how to run examples in PhysiCell-X
|
|
|
|
|
|
|
|
$^2$: An MPI Cartesian Topology is a *virtual* arrangement of processes.
|
|
|
|
$^2$: An MPI Cartesian Topology is a _virtual_ arrangement of processes.
|
|
|
|
|
|
|
|
# References:
|
|
|
|
[1] A. Ghaffarizadeh, S. H. Friedman, and P. Macklin. BioFVM: an efficient, parallelized diffusive transport solver for 3-D biological simulations. Bioinformatics, 32(8):1256–8, 2016. doi: 10.1093/bioinformatics/btv730. URL [http://dx.doi.org/10.1093/bioinformatics/btv730](http://dx.doi.org/10.1093/bioinformatics/btv730).
|
|
|
|
|
|
|
|
[2] A. Ghaffarizadeh, R. Heiland, S. H. Friedman, S. M. Mumenthaler, and P. Macklin. PhysiCell: an open source physics-based cell simulator for 3-D multicellular systems. PLoS Comput. Biol., 14(2):e1005991, 2018. doi: 10.1371/journal.pcbi.1005991. URL [http://dx.doi.org/10.1371/journal.pcbi.1005991](http://dx.doi.org/10.1371/journal.pcbi.1005991).
|
|
|
|
\[1\] A. Ghaffarizadeh, S. H. Friedman, and P. Macklin. BioFVM: an efficient, parallelized diffusive transport solver for 3-D biological simulations. Bioinformatics, 32(8):1256–8, 2016. doi: 10.1093/bioinformatics/btv730. URL <http://dx.doi.org/10.1093/bioinformatics/btv730>.
|
|
|
|
|
|
|
|
\[2\] A. Ghaffarizadeh, R. Heiland, S. H. Friedman, S. M. Mumenthaler, and P. Macklin. PhysiCell: an open source physics-based cell simulator for 3-D multicellular systems. PLoS Comput. Biol., 14(2):e1005991, 2018. doi: 10.1371/journal.pcbi.1005991. URL <http://dx.doi.org/10.1371/journal.pcbi.1005991>.
|
|
|
|
|
|
|
|
[3] A. Kapoulkine. pugixml: Light-weight, simple and fast XML parser for C++ with XPath support, 2016. URL [https://github.com/zeux/pugixml](https://github.com/zeux/pugixml).
|
|
|
|
\[3\] A. Kapoulkine. pugixml: Light-weight, simple and fast XML parser for C++ with XPath support, 2016. URL <https://github.com/zeux/pugixml>.
|
|
|
|
|
|
|
|
[4] G. Saxena, M. Ponce-de Leon, A. Montagud, D. Vicente Dorca, and A. Valencia. BioFVM-X: An MPI+
|
|
|
|
OpenMP 3-D Simulator for Biological Systems. In International Conference on Computational Methods
|
|
|
|
in Systems Biology, pages 266–279. Springer, 2021. URL [https://link.springer.com/chapter/10.1007/978-3-030-85633-5_18](https://link.springer.com/chapter/10.1007/978-3-030-85633-5_18). |
|
|
\ No newline at end of file |
|
|
|
\[4\] G. Saxena, M. Ponce-de Leon, A. Montagud, D. Vicente Dorca, and A. Valencia. BioFVM-X: An MPI+ OpenMP 3-D Simulator for Biological Systems. In International Conference on Computational Methods in Systems Biology, pages 266–279. Springer, 2021. URL <https://link.springer.com/chapter/10.1007/978-3-030-85633-5_18>. |
|
|
\ No newline at end of file |